BioJava:CookBook:PDB:align
How can I calculate a structure alignment?
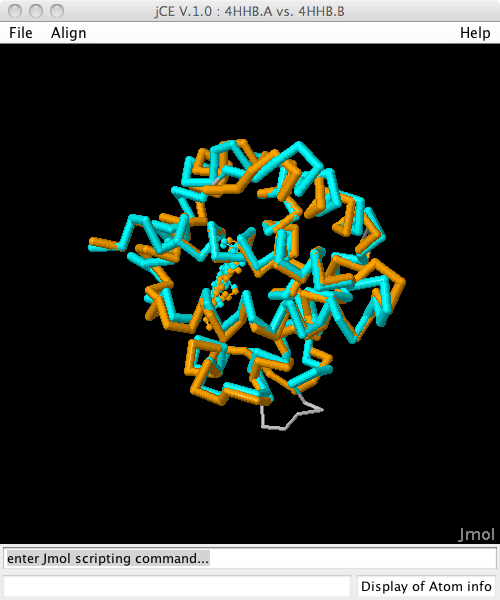
Structure alignment of Hemoglobin Alpha and Beta chain (4hhb.A vs 4hhb.B)
What is a structure alignment?
Structural alignment attempts to establish equivalences between two or more polymer structures based on their shape and three-dimensional conformation. In contrast to simple structural superposition (see below), where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.
For more info see the Wikipedia article on protein structure alignment.
Alignment with Combinatorial Extension (CE) and FATCAT
The structure alignment possibilities of BioJava are going to be greatly enhanced in the upcoming BioJava 3 release. It provides a BioJava port of the Combinatorial Extension algorithm (CE) as well as of the FATCAT algorithm. Thanks to P. Bourne, Yuzhen Ye and A. Godzik for granting permission to freely use and redistribute their algorithms. The documentation how to use these is available from
BioJava:CookBook:PDB:CE_Algorithm
BioJava:CookBook:PDB:FATCAT_Algorithm
Combinatorial Extension with Circular Permutations
Alignment with BioJava 1.7
BioJava 1.7 contains an unpublished structure alignment algorithm. For more information on it, please see here:
BioJava:CookBook:PDB:BioJava_Algorithm
Structure superposition
Also know as “overlaying” is a process of fitting two (equivalent) sets of Atoms onto each other. This can be useful e.g. to overlay two different structural models of the same protein. BioJava also provides a tool for this. (See the SVDSuperimposer.html javadoc.
Where is it being used ?
The protein structure modules of BioJava are used on the RCSB PDB web site for the Protein Comparison Tool.
Interesting test proteins
- 1cdg.A vs 1tim.A . A TIM barrel aligned with a multi domain protein that contains a TIM barrel. While default CE and FATCAT (rigid) parameters only manage to find 3/4 of the barrel aligned, setting the Maximum Gap Size parameter in CE to unlimited (-1) allows it to find the whole barrel. (Example - CE with default gap size)
- 4hhb.A vs. 4hhb.B See how the visualisation tools in BioJava can also display a superposition of the ligands in an alignment. (Example)
- 1a64.A vs. 1hng.B . Domain swapping. In this example the similarity between the two chains can best be found using the FATCAT-flexible algorithm (Compare these examples: FATCAT-flexible vs. CE
- 1vhr.A vs 2ihb.A. Circular permutation. Can be detected when running CE in the circular permutation mode.
See Also
- The BioJava Tutorial on Structure Alignment